Although FOP disease, also known as fibrodysplasia ossificans progressiva, sounds like a clinical term taken from a scholarly medical journal, its implications are incredibly human. The illness reshapes life, bone by unwanted bone, rather than merely limiting it. People who are born with this uncommon genetic mutation usually don’t know what’s in store for them a body that will literally imprison itself in a second skeleton. Imagine your ligaments, muscles, and tendons slowly becoming bone—not in a symbolic sense, but as a result of an inexorable biological process that defies all orders to halt. Unquestionably poetic in its symmetry, this metamorphosis is painfully real and devastating in day-to-day existence. Moments of healing can be transformed into monuments of suffering when a simple vaccination or a stubbed toe serves as the catalyst for a lifetime flare-up, replacing soft tissue with hard bone.
Most cases are diagnosed after patients have already had preventable procedures and injuries, such as biopsies, which can greatly worsen the condition. The cause is a mutation in the ACVR1 gene, which functions similarly to a malfunctioning light switch in that it will not turn off once it is turned on, sending signals to the body to grow bone in tissues where it is not supposed to be. Heterotopic ossification, which traps the body in itself, is the result of this tiny but disastrous error. The first indications, such as malformed big toes at birth or minor skeletal abnormalities in infancy, are frequently disregarded. However, as the illness worsens, particularly after trauma, it changes quickly, freezing limbs, immobilizing joints, and eventually making breathing, speaking, and eating difficult.
| Key Information on FOP Disease | Details |
|---|---|
| Full Name | Fibrodysplasia Ossificans Progressiva |
| Gene Mutation | ACVR1 (Activin A receptor, type I) |
| Inheritance Pattern | Autosomal dominant (but usually a new mutation) |
| First Symptoms | Malformed big toes, stiff neck or back, flare-ups after injury |
| Main Risk Factors | Trauma, surgery, intramuscular injections |
| Treatment Options | Palovarotene (Sohonos), gene-targeted antisense therapy (experimental) |
| Prognosis | Progressive immobility; normal cognitive development |
| Support Organization | International FOP Association (IFOPA) |
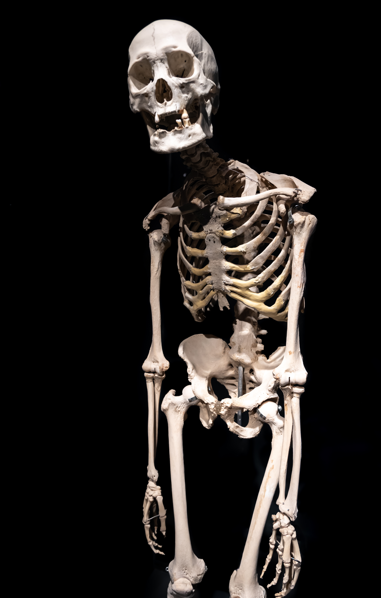
The illness has given rise to a grudging group of silent heroes. One of the most influential people was Harry Eastlack, who was only able to move his lips by the time of his death at the age of 39. As a potent, physical reminder of what FOP does to the body, his skeleton—donated to science—is on exhibit at the Mütter Museum in Philadelphia. The disease’s rarity and devastation are further highlighted by strikingly similar cases like Carol Orzel’s, which is also on display at the museum. These people stand out not only for their clinical rarity but also for the unwavering bravery with which they faced a life that was gradually closing in on them.
Considerable progress has been made in the management of FOP in recent years. During flare-ups, palovarotene, which is marketed under the trade name Sohonos, has demonstrated especially positive results in preventing the formation of new bone. It stops the ossification cascade by focusing on the retinoic acid pathway, but it doesn’t repair damage that has already occurred. Although it is not a cure, this treatment is incredibly effective in giving patients a window of relief by delaying the progression and giving them valuable time. Furthermore, in laboratory settings, antisense therapies that specifically silence the mutated gene exhibit promise, opening up a new treatment option for autosomal dominant diseases like FOP.
Pharmaceutical firms are creating customized strategies through strategic alliances with rare disease research groups that were unimaginable just ten years ago. Trials investigating ways to prevent the pathological activation of the ACVR1 receptor have been led by Ipsen and Regeneron. The foundation for other musculoskeletal disorders involving undesired bone growth is being laid by these studies, which are not limited to FOP. Researchers find that FOP’s specificity makes it an exceptionally clean model for examining how the body misinterprets repair signals, transforming well-meaning healing into an uncontrollable ossification train.
FOP has no effect on cognitive development or intelligence, despite its terrible course. The disease is especially cruel because of this stark contrast to the physical toll it takes. Children who are intellectually active may eventually be deprived of their own physical freedom, which is a distressing experience for both patients and their families. As bone covers their bodies, many experience a significant change in their mobility as well as in how they interact with the outside world, depending more on eye movements, facial expressions, and assistive technology.
The risk of routine medical procedures is what makes FOP even more difficult. Minor surgeries, routine dental work, or intramuscular injections can change a person’s life. The way care is provided has had to change as a result of this; non-invasive methods are now given priority, and patients are frequently encouraged to carry medical alert cards in order to avoid emergency procedures that might worsen their condition. Mobility aids and specialized gear are not only helpful in this situation; they are vital survival tools.
Surprisingly, FOP patients frequently develop into ardent advocates who inform the public and medical community. People from all over the world connect to exchange treatment updates, flare-up management strategies, and emotional support through advocacy platforms such as the International FOP Association (IFOPA). Despite crippling symptoms, their combined fortitude is motivating a new generation of researchers and caregivers who are better prepared than ever to make a significant impact.
Precision medicine might be the solution in the years to come. The potential to silence or fix the defective ACVR1 gene before ossification ever starts is a tantalizing prospect presented by advances in gene editing, especially CRISPR-Cas9. This condition may one day be treatable or even preventable if detected early enough, possibly even during pregnancy. Until then, the battle goes on—not only in labs or hospitals, but also in the daily lives of people who have a rare and mercilessly cruel illness.